Team Christian Stolte LEAD UX DESIGN + VISUAL DESIGN Avinash Abhyankar BIOINFORMATICS LEAD Kevin Shi, Nathaniel Novod SOFTWARE ENGINEERING Nina Lapchyk QA MANAGEMENT Kanika Arora, Minita Shah CANCER BIOINFORMATICS Hemali Phatnani, Toby Bloom PRINCIPAL INVESTIGATORS

The beginning of chromosome 1 in a VCF file of genomic variants, shown in a spreadsheet...
Let’s say you are a researcher who received money from the ice bucket challenge and you want to investigate patients with ALS. You sequence the genomes of a cohort of patients.
From that you get a file for each patient that lists the differences between a human reference genome and that individual—3.5 million variants, on average.
You can look at that list in a spreadsheet, shown left —frustrating, to say the least. You realize that you need computational help sorting these variants, assigning them to genes, predicting their effect. The challenge becomes bigger when you want to consider not just one, but all your patients:
• find out which variants they have in common,
• filter out variants that occur in healthy individuals,
• investigate which variants occur together more frequently with certain symptoms.
You search the scientific literature for tools to do these tasks, and find some promising descriptions. At closer inspection, most of the software packages for individual genome analysis turn out to be command line applications without a graphic user interface, written in a variety of languages: Python, R, Java, and more. Their installation can be difficult because they depend on a host of other packages. Their output is, most often, another list. Visualizing the results is up to you.
Looking for tools to tackle cohorts, you find some online resources that present data from public projects and include visual interfaces to explore the data. You could use them as reference or to take the place of healthy controls. But none of those resources allow you to import and integrate your own data. You are stuck.
To be realistic, no researcher would embark on such a project on their own. They would assemble a team of people with expertise in the required areas, and split the available funding between paying their salaries and generating data. That often means recruiting fewer patients, resulting in a study with reduced statistical power.
Fixing a workflow bottleneck
Biology is enormously complex. To make progress, scientists hone in on specific aspects of biological processes, and accumulate deep knowledge about narrow subject areas. Thus, each biologist has their own view of the human organism. To tackle a poorly understood disease such as ALS, it would take many different researchers, who all had different questions they wanted to ask of the data. Each question meant a different algorithm to write, a different angle from which to approach the data...
If the solution were to pair up each biologist with a bioinformatician, we had a problem: based on open positions in the US, there was only one bioinformatician for every eight biologists. That kind of skewed proportion pointed out a serious obstacle on the path to new discoveries.
If we couldn’t magically create bioinformaticians, how could we make better use of their time?
We wanted to empower regular biologists to explore data on their own, so they could use their knowledge and scientific intuition to generate new hypotheses and get initial confirmation. They would then follow up only the best leads with their computational colleagues for thorough analysis.
Design decisions to support exploration
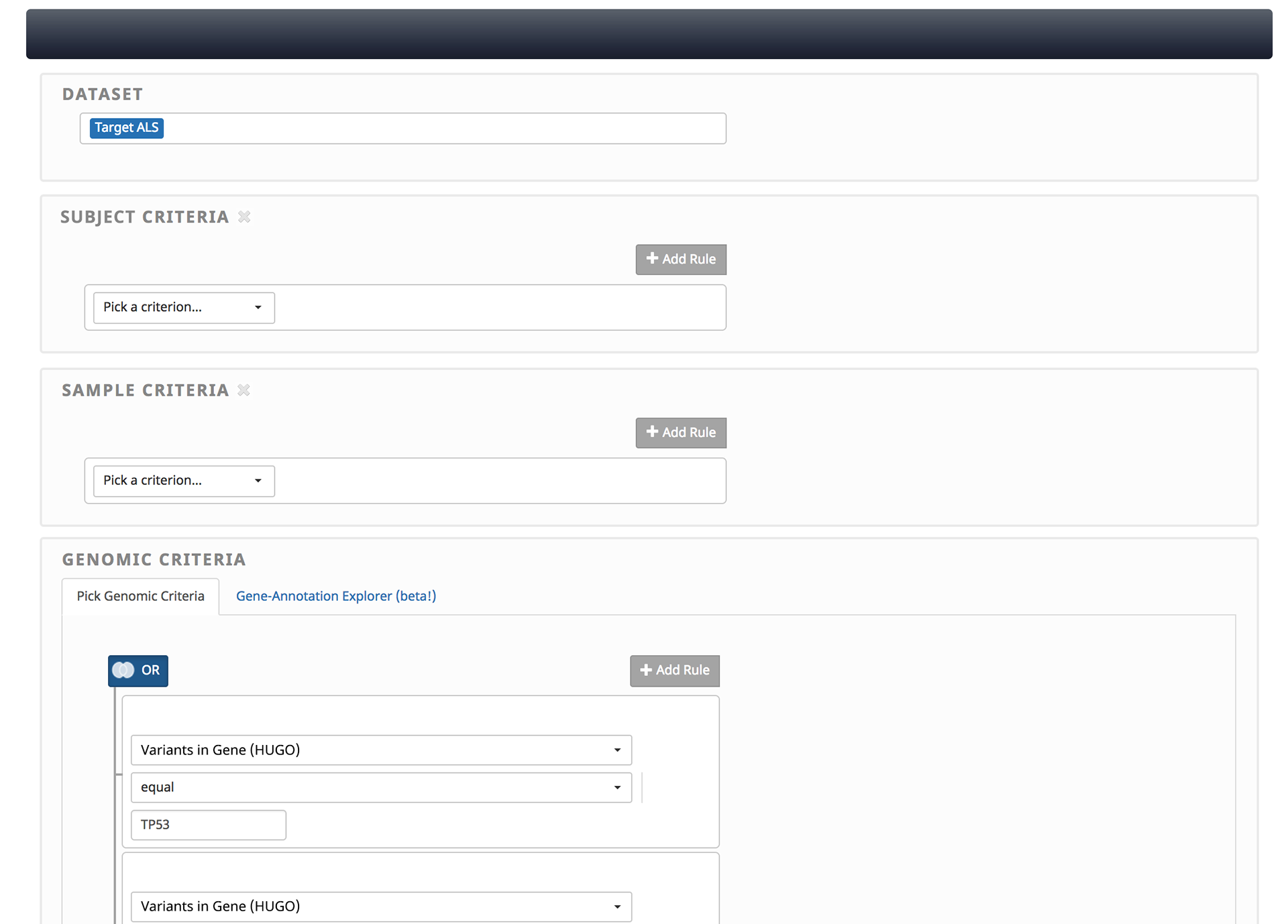
1. Query
Every inquiry begins with a question about the data, such as “show me people with ALS who have variants in my favorite gene”—that will be your cohort to explore. In MetroNome, this question can be formed with a query builder that uses drop-down menus to build “rules” to select that cohort.
Starting with one or more datasets, you can then use a combination of criteria to define the resulting list of individuals. Criteria can be chained together using the intersection of result sets (AND) or the union (OR).
2. Visualize results
We show the results in linked interactive diagrams that present data in a biologically relevant context. RNA expression is shown for the selected gene in a diagram that maps expression values to anatomical regions of the brain and spinal cord (top right).
MetroNome displays variants (bottom) in gene diagrams that include functional domains for the protein transcript to serve as landmarks for biologists.
Side-by-side comparisons of two cohorts let users spot differences and unique attributes.
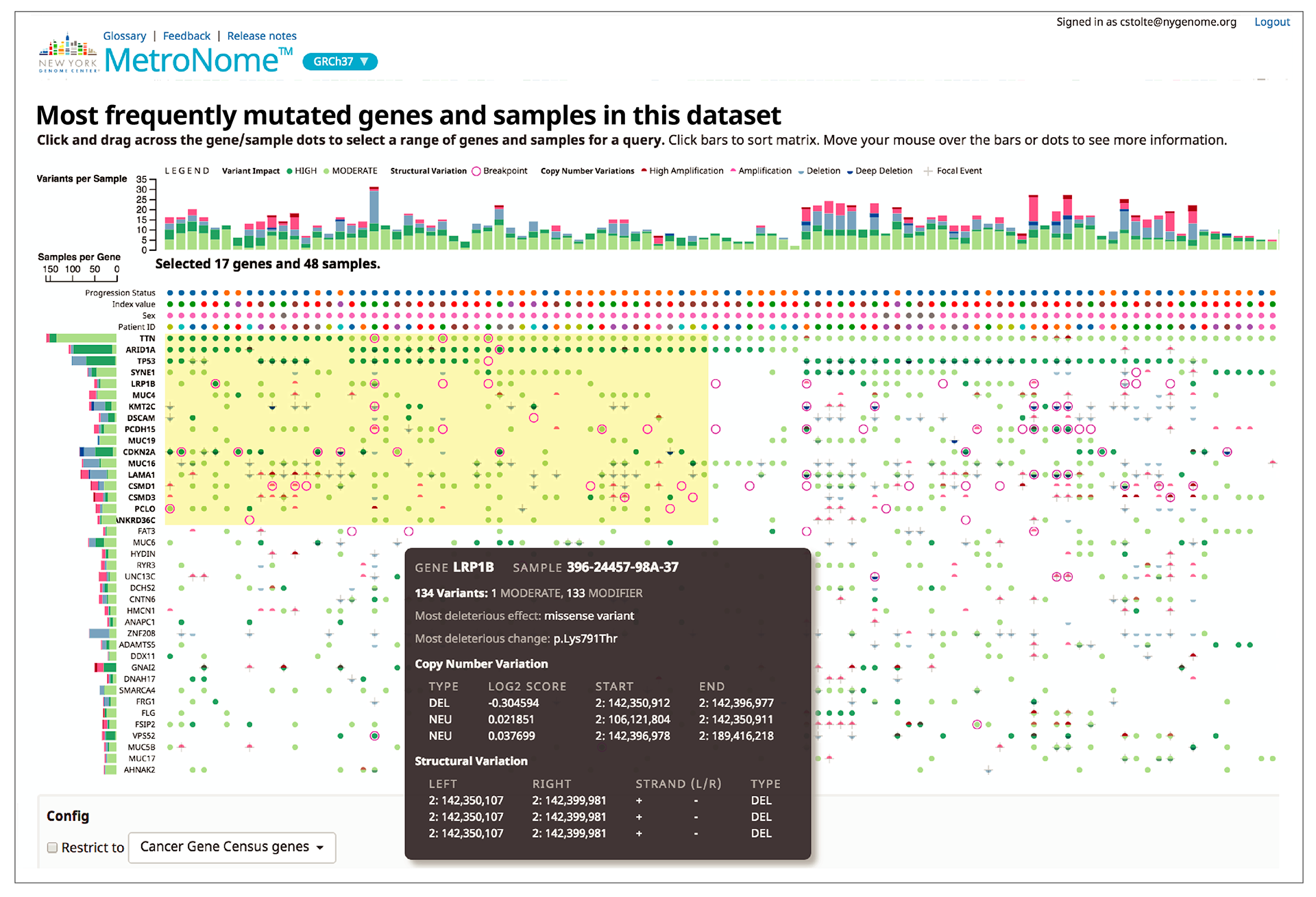
The flexible UI lets you choose from 10 customizable panels for a wide a variety of views into the data.

This sortable matrix of genes (rows) and samples (columns) lets researchers explore the data for different types of mutations.
A subset of samples and genes can be selected to launch a new query.
3. Download results
Once you have found an interesting cohort or set of variants, you can download the results as CSV files. The list of subject IDs will give you a starting point for a thorough follow-up analysis.
