Team Christian Stolte UX DESIGN + VISUAL DESIGN Seán O’Donoghue DESIGN + BIOINFORMATICS LEAD
Kenny Sabir, Julian Heinrich SOFTWARE ENGINEERING Seán O’Donoghue PRINCIPAL INVESTIGATOR

This still frame from Chris Hammang’s animation Alzheimer’s Enigma shows the final model for the overall structure of the amyloid precursor protein (APP), rendered in yellow, which was assembled using Aquaria.
Proteins are the building blocks of life. They form the molecular machinery involved in almost every process in a living organism.
Each protein is made up of a sequence of amino acids. This string of amino acids folds into a three-dimensional shape, or 3D structure, which is critical for its function. Ever since the discovery of the DNA double helix, biologists have been aware that atomic-scale threedimensional (3D) structures can provide significant insight. We know 168 million protein sequences*, yet we only have experimentally determined structures for a little more than 160,000 of them**—that is less than one in a thousand. Computational prediction of 3D structure from sequence is still imperfect.
Making it easier to find and view protein structures
Using a database of computed similarity between sequences, Aquaria is a web resource that shows known structures for proteins of similar sequence. Using this approach, we matched more than 1 million extra protein sequences with a structure, compared to the contents of the PDB.
The user interface and the visualizations had to address:
• quality of the match
• alignment of the sequence, relative to the query protein
• clustering of results (sometimes hundreds)
• user choice of structure
• display of annotated features
• interactive connection between sequence and structure
• display of additional protein information
• alignment of the sequence, relative to the query protein
• clustering of results (sometimes hundreds)
• user choice of structure
• display of annotated features
• interactive connection between sequence and structure
• display of additional protein information
The primary audience for this resource were researchers in the life sciences without structural biology background.
The goal was to provide easy access to 3D structures, while maintaining an interactive connection between sequence and structure.
Other considerations were to follow established conventions for the display of structures. As the default view, we chose the Ribbon display, because it provides secondary structure elements and makes it easy to follow the sequence through the 3d structure.
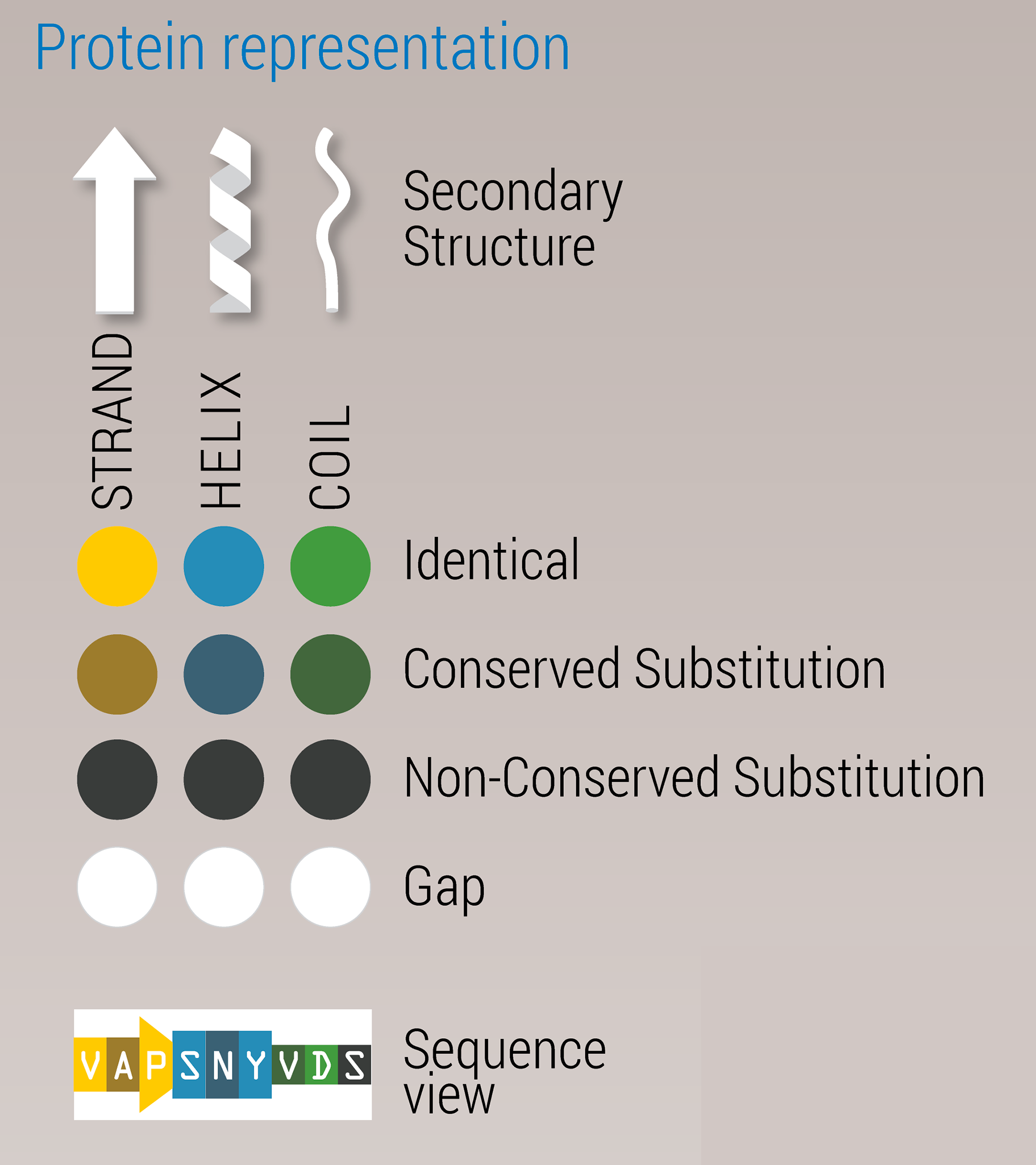

The color schemes that were prototyped to convey secondary structure elements, amino acid composition, and conservation: when seen in color blind simulation, blue and yellow hues are best preserved; contrast is best in the bottom row.
When proteins fold into their final shape, they follow three patterns, called secondary structure: the corkscrew-like ‘coils’, the flat ‘sheets’, and free-form ‘loops’. We decided on a color scheme for the structures that was colorblind-safe.
The three secondary structure elements are also represented by different shapes in the sequence view.
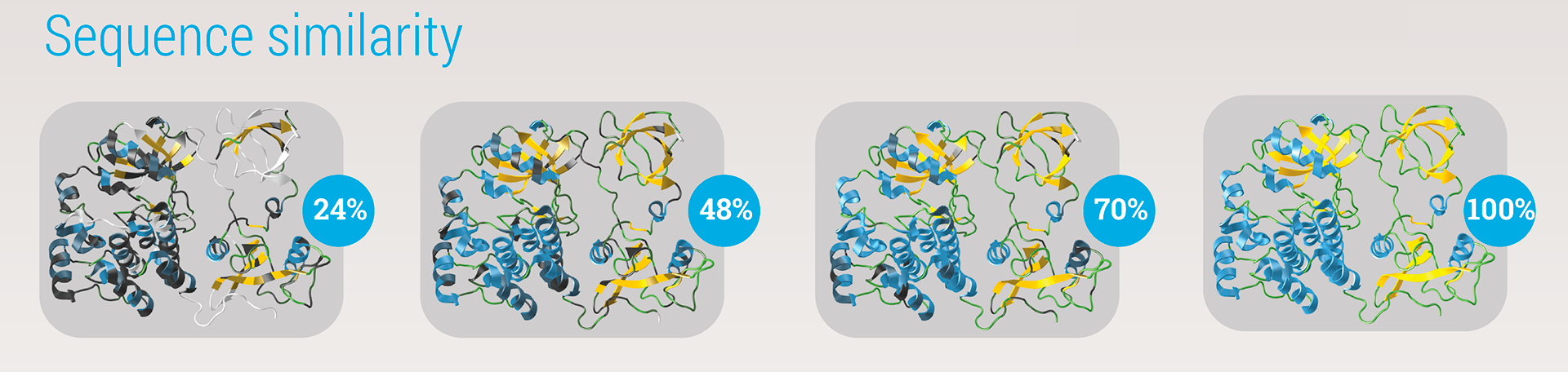
The quality of the match for the entire structure is expressed in a percentage. Per amino acid, a mismatch is color-encoded by increasing darkness. A poorly matched structure would therefore appear to have a lot of dark spots.

The Aquaria user interface comprises five panels: a 3D view (A) shows the currently selected 3D structure with various rendering modes using the same colour scheme as applied for all structures and aggregates (E) that match a given user query entered through the search field (B). A white background is used to visually connect the sequence being rendered in the 3D view and its cluster in the matching sequences panel (E). Panels on either side give information about the Uniprot entry corresponding to the query (B) and its function (C), as well as details of the structure (D) being shown in the 3D view.
Ease of search: users can input a protein name, e.g., ‘insulin’, or a UniProt identifier, and an auto-complete function would suggest matches in the database. We decided to show the structure with the longest alignment and best match in the 3D view in the center of the window. Users can then dig into the search results (below) and select alternate structures for display. To the left and right we display information about the protein and the structure, sourced from Uniprot and PDB.
We had to devise a compact way to display the results, which often number in the hundreds. We decided to cluster results into groups, arranged by sequence alignment. The glyph representing a group shows the shared secondary structure elements. The color indicates the quality of the match. A numeric label shows the size of the cluster. Users can drill down into these groups. When expanded, a fan of subgroups opens up, sorted by third-order conformation: monomer, dimer, etc. These subgroups expand in turn to show individual structures, sorted by quality.
Next to the tab for results is a tab for annotated features. When selected, users see an array of annotations that can be selected to color the structure.
* as of August 10, 2019, UniProt.org held sequences for 560,537 manually annotated and reviewed proteins (SwissProt), and 167,761,270 that were automatically annotated and not reviewed (trEMBL).
** wwpdb.org had a total of 161,554 structures deposited as of Aug 10, 2019.